
We are going to start our interpretation of a variant calls by paraphrasing the description from the Broad Institute:
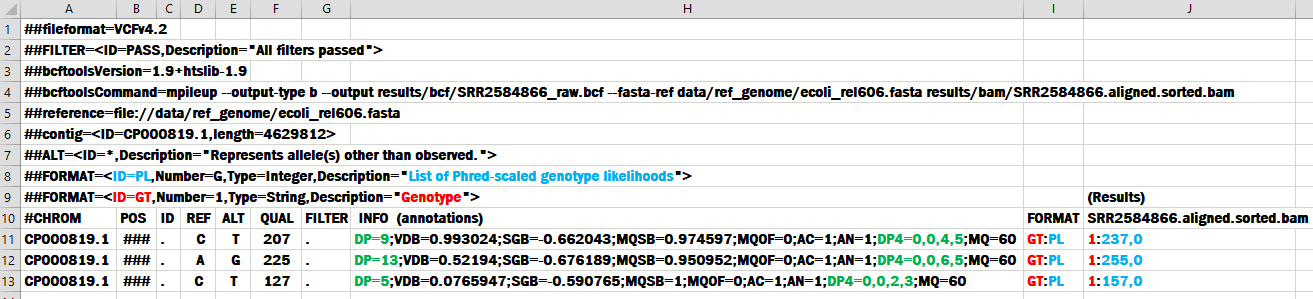
The differences between .VCF files makes this topic harder to grasp. We will spend a little time on the meanings of some metrics to help keep your bioinformatics workflow operating. First we present an image to help understand the locations and relationships between the headers and the records:
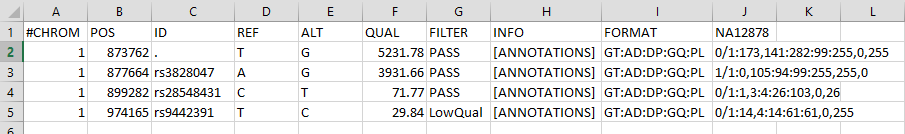
Here’s an image of different SNP alleles from the Lecture (and described below) opened in a spreadsheet without the headers:
Note that a GT allele assignment of 0/0 (hom-ref) essentially means the reads match the REF genome. Which is
by definition NOT a variant. When that occurs, the first of three PL values are sometimes omitted from VCF files.
Remember this when you go back to our SRR2584866_final_variants.vcf file in the lesson and the -v flag in
the bcftools call command!
When all three PL values are shown they represent genotype calls for hom-ref,het,hom-alt (REF,HET,ALT)
The result for any hom-ref allele, when all three PL values are always shown, will be 0,<value>,<value>
The result when hom-ref alleles are not shown in VCF files is the two values listed
for a PL metric, are: <value>,0 (hom-alt) or 0,<value> (het).
However, the hom-ref value can help define variants, or indicate problems in the
variant “call” when a hom-alt is shown as: <value>,0,<value> or <value>,<value>,0
If this seems confusing, you are not alone! The max <value> (the lowest probability) is 255 representing 10^(-25.5)
and 0 means 10^(-0) = 1. So 0 is the most certain, and 255 is the least certain. Some examples are below:
Wait! What about haploid vs ploidy?
This is a great question, and there are more details about ploidy actively being developed. For now, the issue is only partially resolved, and when mapping reads or contigs to a reference genome the reference genome (e.g. human) is usually represented as haploid. For now, some software try to infer the ploidy level, and other software (e.g. BWA-MEM) actively work to use ploidy in the analyses. This is beyond the scope of our lesson, but you can represent haploid or diploid variants as
REF/ALT(0/0,0/1,0/2, etc.) and call genotypes as0,1,2… etc.Note: This isn’t a trivial problem as in humans the autosomes are diploid, the sex chromosomes are (mostly) haploid, and mitochondrial genomes are polyploid!
Interesting examples of this problem are discussed at https://www.biostars.org/p/348867/ and https://galaxyproject.org/tutorials/var_hap/
Example 1:
The genotype information for a RESULTS column named NA12878, at Chromosome 1 position 899282
(Where it says “<snip>” we just omitted some annotation).
1 899282 rs28548431 C T <snip> GT:AD:DP:GQ:PL 0/1:1,3:4:26:103,0,26
At this SNP site, the called genotype GT is het (heterozygous) GT = 0/1, which corresponds to the alleles C/T.
Yes, this can be confusing, because one might think the “GT” is the genotype
G/T, butGTis the “TAG” ABBREVIATION for the “GenoType” short name metric.
The GQ tag
Notice the confidence indicated by GQ = 26 isn’t very good, largely
because there were only a total of 4 reads at this site (DP = 4), 1 of which matched the REF ( = had the reference base)
and 3 of which matched the ALT ( = had the alternate base) as indicated by AD = 1,3. In very simple terms the GQ is defined as:
“The difference between the second lowest PL and the lowest PL (the lowest PL is always 0)”
The lack of certainty in calling this allele also shows up in the PL
fields which are 103,0,26.
The reason for this allele call is because the hom-ref allele PL value
is 103 or 10^(-10.3) which is close to 0 while
the PL for the ALT allele hom-var is PL = 26 (which corresponds to a likelihood
of 10^(-2.6), or 0.0026) so it is unlikely but possible).
CONCLUSIONS: We can only conclude that the subject is definitely not hom-ref (homozygous with
the reference allele) and actually has a slim chance of being hom-var
(homozygous with the variant allele)! The call for position 899282 as het is probably correct,
but there’s a slight chance that the genotype assignment of het (GT=0/1)
is incorrect, therefore more coverage is needed at this site.
Optional Example 2:
Now let’s try explaining the example from the Broad institutes example at position 873762.
We’ll use our own words:
First, recognize that a “genotype” (i.e. the GT metric) can have
at least three possibilities displayed as REF/ALT (or sometimes REF,ALT):
- GT (0/0) “homozygous with the REF allele” (This basically means there is no variant)
- GT (0/1) “heterozygous at REF allele”
- GT (1/1) “homozygous at ALT allele” (This basically means the base in the REF genome is the variant)
1 873762 . T G <snip> GT:AD:DP:GQ:PL 0/1:173,141:282:99:255,0,255
The called genotype is T/G, and the genotype metric GT has the value: 0/1 = a heterozygous allele (het)
The confidence metric GQ has a value of 99 which is the MAXIMUM value you can assign (all GQ confidence levels are capped at 99)
The total “raw” reads depth metric DP has a value of 282 reads.
The allelic reads metric AD shows there were 173 reads matching the REF genome and 141 reads matching the ALT genome.
- Note that of 282 reads, 141 were ALT, for a ratio of exactly 0.5
- This tells us the locus has about a nearly perfect heterozygosity score for a diploid genome (141/282 = 50%)
- Also, we know some reads were not used for the
GTmetric because 173 + 141 = 314 reads, which is greater than theDP“total” reads metric (282).
The likelyhood metrics are shown as PL values for each type of allele are 255,0,255.
Now we know that the different VCF file options in each column have assigned positions, divided by colons, and that each metric can have multiple values divided by commas. Also, these will vary depending on which software generated the file and may not report some metrics values.
PL Metric take home for this example
- PL@position-1 (“Likelihood of homozygous with the REF allele”) = 255. This corresponds to 10^(-25.5) a very small number
- PL@position-2 (“Likelihood of heterozygous at REF allele”) = 0. The assigned allele is always normalized to zero (the maximum likelyhood)
- PL@position-3 (“Likelihood of homozygous at ALT allele”) = 255. This corresponds to 10^(-25.5) a very small number
CONCLUSIONS: Position 873762 is definitely not hom-ref or hom-var, and must be het. Also the confidence metric GQ is maxxed out at 99 so
the certainty is as high as it can get!
Therefore:
- We’re very sure there’s a variant at this site, and
- There’s not much chance the genotype assignment is incorrect, and
- The sample is het (heterozygous) for T/G at this locus.
Now that it’s all very clear, we can go back to the variant calling workflow page.