
Introduction to Shell Loops
Loops are key to productivity improvements!
They allow us to automate the execution of repetitive commands.
Similar to wildcards and tab completion, using loops also reduces
the amount of typing (and typing mistakes).
In Genomics, loops are helpful when performing operations on
groups of sequencing files, such as unzipping or trimming multiple
files. We will use loops for these purposes in subsequent analyses, but
will cover the basics of them for now.
Writing for loops
When the shell sees the keyword for, it knows to repeat a command
(or group of commands) once for each item in a list.
Each time the loop runs (called an iteration), an item in the iterated list is assigned to the variable, and the commands inside the loop are executed, before moving on to the next item in the list.
Inside the loop, we call for
the variable’s value by putting $ in front of the variable’s name
(which we enclose in curly brackets). The $
tells the shell interpreter to substitute the variable’s value for
the variable’s name,
rather than treat it as text or a command.
Temporarily Changing the Prompt
Most of you probably have a “$” as your terminal prompt. Some of you may have the greater-than symbol “>”. In either case, this could cause confusion as we are just starting to use loops. So we are going to change our prompt to a question mark followed by a space ”? “. This is only temporary, and you don’t have to do this if you are pretty sure you won’t get confused when using the “$” for variables in loops.
To change your prompt from “$ “ to “? “ just type:
PS1="? "If you want to change it back, just type:
PS1="$ "If you want a prompt that shows your current working directory type:
PS1="[\\w] ? "
Now let’s write a for loop to show us the first five lines of
Nelle’s data files.
Starting from the data-shell directory, cd to the data/ directory
$ cd data/
Check what is in this directory:
? ls -F
amino-acids.txt animals.txt morse.txt planets.txt sunspot.txt
animal-counts/ elements/ pdb/ salmon.txt
change into the pdb directory and look around
? cd pdb
? ls -F
aldrin.pdb glycol.pdb pentane.pdb
ammonia.pdb heme.pdb piperine.pdb
ascorbic-acid.pdb lactic-acid.pdb propane.pdb
benzaldehyde.pdb lactose.pdb pyridoxal.pdb
camphene.pdb lanoxin.pdb quinine.pdb
cholesterol.pdb lsd.pdb strychnine.pdb
cinnamaldehyde.pdb maltose.pdb styrene.pdb
citronellal.pdb menthol.pdb sucrose.pdb
codeine.pdb methane.pdb testosterone.pdb
cubane.pdb methanol.pdb thiamine.pdb
cyclobutane.pdb mint.pdb tnt.pdb
cyclohexanol.pdb morphine.pdb tuberin.pdb
cyclopropane.pdb mustard.pdb tyrian-purple.pdb
ethane.pdb nerol.pdb vanillin.pdb
ethanol.pdb norethindrone.pdb vinyl-chloride.pdb
ethylcyclohexane.pdb octane.pdb vitamin-a.pdb
So we have found Nelle’s stash of metabolite data that was passed down to her from a previous graduate student. When we check to see if the 48 files are all similar we see they are not:
? wc -l *.pdb
30 aldrin.pdb
7 ammonia.pdb
24 ascorbic-acid.pdb
etc...
34 tyrian-purple.pdb
23 vanillin.pdb
10 vinyl-chloride.pdb
55 vitamin-a.pdb
1808 total
So what do the files look like? We can look at each
one individually, but what if there are 6000
files? What if the files were much larger (e.g. 3Gb each)? We
know we can look at the beginnings of any file using the head
command so let’s try that first:
? head -n 5 aldrin.pdb
COMPND ALDRIN
AUTHOR DAVE WOODCOCK 97 08 06
ATOM 1 C 1 -0.888 0.654 -1.753
ATOM 2 C 1 0.276 1.404 -1.135
ATOM 3 C 1 1.381 0.453 -0.730
These files look a little complex. To see if they are all generated
by the same person and have the same date, or additional
information, we will generate a short summary
of each file using a for loop. We could do this a different way,
but this prepares us for later lessons where we will use much bigger files.
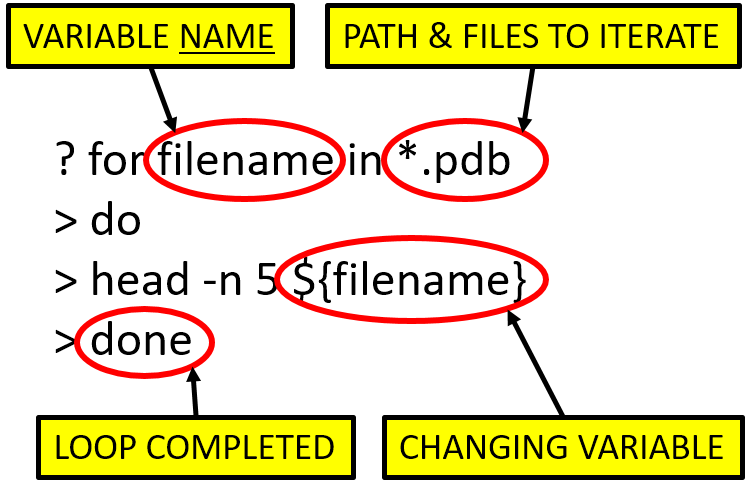
The image below gives the basic anatomy of our “loop”
Now let’s type the loop. (remember don’t type in the “?” sign, that’s the prompt):
? for filename in *.pdb
> do
> head -n 5 ${filename}
> done
The for loop begins with the for command which includes:
for <variable> in <file/group to iterate over>. This multi-part
command is sometimes called a “formula”.
Notice that the word filename is designated
as the variable to be used with each iteration. In our case all
files ending with .pdb in the current working directory (e.g. aldrin.pdb, ammonia.pdb, etc.)
will be substituted for filename. When we hit the Enter key
we notice the prompt has changed to a > symbol. This means the shell
recognized the for command and is waiting for us to finish our loop.
The next line of the for loop is do
(meaning “do” something). This is followed by a line describing
what we want to execute (i.e. what to “do”).
We are telling the loop to print the first
five lines of each variable (head -n 5 ${filename}) as we iterate
over all the files. To be clear, the value of the
variable $[filename} is the actual name of each *.pdb file.
The loop generates the commands:
head -n 5 aldrin.pdb
head -n 5 ammonia.pdb
head -n 5 ascorbic-acid.pdb
(etc.)
Finally, the word done tells the shell we have finished the loop.
After executing the loop, you should see the first five lines of
all .pdb files printed to the terminal.
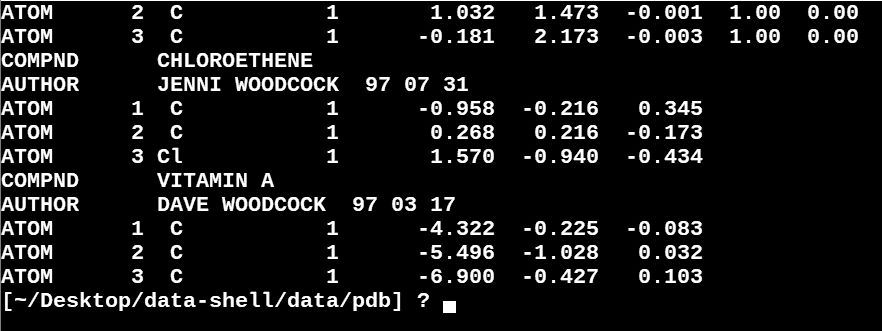
But again, what if there are 6000 files? Let’s create a loop that will save this information to a file we can look at later.
? for filename in *.pdb
> do
> head -n 5 ${filename} >> pdb_info.txt
> done
Note that we are using >> to append the text to our pdb_info.txt file.
If we used >, the pdb_info.txt file would be rewritten
every time the loop iterates, so it would only have text from the last
variable used. Instead, >> continuously adds to the end of the file.
Use cat to verify the pdb_info.txt file looks like the screen output
you observed when running the loop without any redirection.
This lesson is very similar to the one we will use for manipulating files in the genomics part of the course. For now, let’s see what Nelle is going to do with loops.