
Learning Objectives
Following this assignment students should be able to:
- practice basic syntax and usage of
forloops- use
forloops to automate function operations- understand how to decompose complex problems
Reading
-
Topics
- Iteration
- Problem decomposition
-
Readings
Lecture Notes
Exercises
Use and Modify with Apply (15 pts)
This is a followup to Use and Modify.
-
Write a function that takes length as an argument to get an estimate of mass values for the dinosaur Theropoda. Use the equation
mass <- 0.73 * length ** 3.63, where 0.73 is a and 3.63 is b. Then usesapply()and the following vector of lengths to get a vector of estimated masses.theropoda_lengths <- c(17.8013631070471, 20.3764452071665, 14.0743486294308, 25.65782386974, 26.0952008049675, 20.3111541103134, 17.5663244372533, 11.2563431277577, 20.081903202614, 18.6071626441984, 18.0991894513166, 23.0659685685892, 20.5798853467837, 25.6179254233558, 24.3714331573996, 26.2847248252537, 25.4753783544473, 20.4642089867304, 16.0738256364701, 20.3494171706583, 19.854399305869, 17.7889814608919, 14.8016421998303, 19.6840911485379, 19.4685885050906, 24.4807784966691, 13.3359960054899, 21.5065994598917, 18.4640304608411, 19.5861532398676, 27.084751999756, 18.9609366301798, 22.4829168046521, 11.7325716149514, 18.3758846100456, 15.537504851634, 13.4848751773738, 7.68561192214935, 25.5963348603783, 16.588285389794) -
Rewrite the function with a and b as arguments, then use
mapply()to determine the mass estimates for the dinosaurs using the following vectors of a and b values for each dinosaur.a_values <- c(0.759, 0.751, 0.74, 0.746, 0.759, 0.751, 0.749, 0.751, 0.738, 0.768, 0.736, 0.749, 0.746, 0.744, 0.749, 0.751, 0.744, 0.754, 0.774, 0.751, 0.763, 0.749, 0.741, 0.754, 0.746, 0.755, 0.764, 0.758, 0.76, 0.748, 0.745, 0.756, 0.739, 0.733, 0.757, 0.747, 0.741, 0.752, 0.752, 0.748)b_values <- c(3.627, 3.633, 3.626, 3.633, 3.627, 3.629, 3.632, 3.628, 3.633, 3.627, 3.621, 3.63, 3.631, 3.632, 3.628, 3.626, 3.639, 3.626, 3.635, 3.629, 3.642, 3.632, 3.633, 3.629, 3.62, 3.619, 3.638, 3.627, 3.621, 3.628, 3.628, 3.635, 3.624, 3.621, 3.621, 3.632, 3.627, 3.624, 3.634, 3.621)
-
Crown Volume Calculation (15 pts)
The UHURU experiment in Kenya has conducted a survey of Acacia and other tree species in ungulate exclosure treatments. Data for the tree data is available here in a tab delimited (
"\t") format. Each of the individuals surveyed were measured for tree height (HEIGHT) and canopy size in two directions (AXIS_1andAXIS_2). Read these data in using the following code:tree_data <- read.csv("http://www.esapubs.org/archive/ecol/E095/064/TREE_SURVEYS.txt", sep = '\t', na.strings = c("dead", "missing", "MISSING", "NA", "?", "3.3."), stringsAsFactors = FALSE)You want to estimate the crown volumes for the different species and have developed equations for species in the Acacia genus:
volume = 0.16 * HEIGHT^0.8 * pi * AXIS_1 * AXIS_2and the Balanites genus:
volume = 1.2 * HEIGHT^0.26 * pi * AXIS_1 * AXIS_2For all other genera you’ll use a general equation developed for trees:
volume = 0.5 * HEIGHT^0.6 * pi * AXIS_1 * AXIS_2-
Write a function called
tree_volume_calcthat calculates the canopy volume for the Acacia species in the dataset. To do so, use an if statement in combination with thestr_detect()function from thestringrR package. The codestr_detect(SPECIES, "Acacia")will returnTRUEif the string stored in this variable contains the word “Acacia” andFALSEif it does not. This function will have to take the following arguments as input: SPECIES, HEIGHT, AXIS_1, AXIS_2. Then run the following line:tree_volume_calc("Acacia_brevispica", 2.2, 3.5, 1.12) -
Expand this function to additionally calculate canopy volumes for other types of trees in this dataset by adding if/else statements and including the volume equations for the Balanites genus and other genera. Then run the following lines:
tree_volume_calc("Balanites", 2.2, 3.5, 1.12)tree_volume_calc("Croton", 2.2, 3.5, 1.12) -
Now get the canopy volumes for all the trees in the
tree_datadataframe and add them as a new column to the data frame. You can do this usingtree_volume_calc()and eithermapply()or usingdplyrwithrowwiseandmutate.
-
Use and Modify with Loops (15 pts)
This is a followup to Use and Modify. It is a partner exercise to Use and Modify with Apply.
-
Write a function that takes length as an argument to get an estimate of mass values for the dinosaur Theropoda (or copy the one you wrote as part of Use and Modify). Use the equation
mass <- 0.73 * length ** 3.63, where 0.73 isaand 3.63 isb. Then use aforloop over the values of the following vector of lengths to print out the estimated masses for each length.theropoda_lengths <- c(17.8013631070471, 20.3764452071665, 14.0743486294308, 25.65782386974, 26.0952008049675, 20.3111541103134, 17.5663244372533, 11.2563431277577, 20.081903202614, 18.6071626441984, 18.0991894513166, 23.0659685685892, 20.5798853467837, 25.6179254233558, 24.3714331573996, 26.2847248252537, 25.4753783544473, 20.4642089867304, 16.0738256364701, 20.3494171706583, 19.854399305869, 17.7889814608919, 14.8016421998303, 19.6840911485379, 19.4685885050906, 24.4807784966691, 13.3359960054899, 21.5065994598917, 18.4640304608411, 19.5861532398676, 27.084751999756, 18.9609366301798, 22.4829168046521, 11.7325716149514, 18.3758846100456, 15.537504851634, 13.4848751773738, 7.68561192214935, 25.5963348603783, 16.588285389794) -
Write a new version of the function that takes
aandbas arguments (or copy the one you wrote as part of Use and Modify), then use aforloop over an index to print the mass estimates for the dinosaurs using the following vectors ofaandbvalues for each dinosaur.a_values <- c(0.759, 0.751, 0.74, 0.746, 0.759, 0.751, 0.749, 0.751, 0.738, 0.768, 0.736, 0.749, 0.746, 0.744, 0.749, 0.751, 0.744, 0.754, 0.774, 0.751, 0.763, 0.749, 0.741, 0.754, 0.746, 0.755, 0.764, 0.758, 0.76, 0.748, 0.745, 0.756, 0.739, 0.733, 0.757, 0.747, 0.741, 0.752, 0.752, 0.748)b_values <- c(3.627, 3.633, 3.626, 3.633, 3.627, 3.629, 3.632, 3.628, 3.633, 3.627, 3.621, 3.63, 3.631, 3.632, 3.628, 3.626, 3.639, 3.626, 3.635, 3.629, 3.642, 3.632, 3.633, 3.629, 3.62, 3.619, 3.638, 3.627, 3.621, 3.628, 3.628, 3.635, 3.624, 3.621, 3.621, 3.632, 3.627, 3.624, 3.634, 3.621) -
Write a new version of the loop from Task 2 that stores the resulting masses in a vector instead of printing them out. Once the loop is finished display the resulting vector.
-
Multiple Files (20 pts)
A colleague wants to do a comparitive analysis of the GC content (i.e., the percentage of bases in a DNA sequence that are either G or C) on data from the PATRIC bacterial phytogenomic database. They want to know the GC content of all of the bacteria in the database and have started working initially with a handful of archaea. They know a little R and wrote the following code to load a single sequence and caculate it’s GC content.
library(ShortRead) library(Biostrings) reads <- readFasta("archaea-dna/A-saccharovorans.fasta") seq <- sread(reads) base_freq <- alphabetFrequency(seq) gc_content <- (base_freq[1, "G"] + base_freq[1, "C"]) / sum(base_freq) * 100The first two lines load a .fasta file using the ShortRead package in Bioconductor. The second two lines determine the frequency of all of the bases in sequence and then calculate the GC content.
Start by installing Bioconductor with the following code (this may take a while, so be patient):
source("https://bioconductor.org/biocLite.R") biocLite("ShortRead") biocLite("Biostrings")Each fasta file contains one long sequence of DNA for an archaea species. The following code loads one sequence file, where
seqis the variable name for the data file:library(ShortRead) reads <- readFasta("archaea-dna/A-saccharovorans.fasta") seq <- sread(reads)Help out your colleagues by downloading the data and looping over the files to determine the GC content for each file. Once downloaded, either unzip the .zip file manually or use the
unzipfunction.Use
[click here for output]list.files(), withfull.namesset to true, to generate a list of the names of all the sequence files. Then create a for loop that uses the above code to read in each sequence file and calculate it’s GC content. Store the resulting values in a data frame with one column with file names and a second column with GC contents.DNA or RNA Iteration (15 pts)
This is a follow-up to DNA or RNA.
Write a function,
dna_or_rna(sequence), that determines if a sequence of base pairs is DNA, RNA, or if it is not possible to tell given the sequence provided. Since all the function will know about the material is the sequence the only way to tell the difference between DNA and RNA is that RNA has the base Uracil ("u") instead of the base Thymine ("t"). Have the function return one of three outputs:"DNA","RNA", or"UNKNOWN".- Use the function and a
forloop to print the type of the sequences in the following list. - Use the function and
sapplyto print the type of the sequences in the following list.
sequences = c("ttgaatgccttacaactgatcattacacaggcggcatgaagcaaaaatatactgtgaaccaatgcaggcg", "gauuauuccccacaaagggagugggauuaggagcugcaucauuuacaagagcagaauguuucaaaugcau", "gaaagcaagaaaaggcaggcgaggaagggaagaagggggggaaacc", "guuuccuacaguauuugaugagaaugagaguuuacuccuggaagauaauauuagaauguuuacaacugcaccugaucagguggauaaggaagaugaagacu", "gauaaggaagaugaagacuuucaggaaucuaauaaaaugcacuccaugaauggauucauguaugggaaucagccggguc")Optional: For a little extra challenge make your function work with both upper and lower case letters, or even strings with mixed capitalization
[click here for output]- Use the function and a
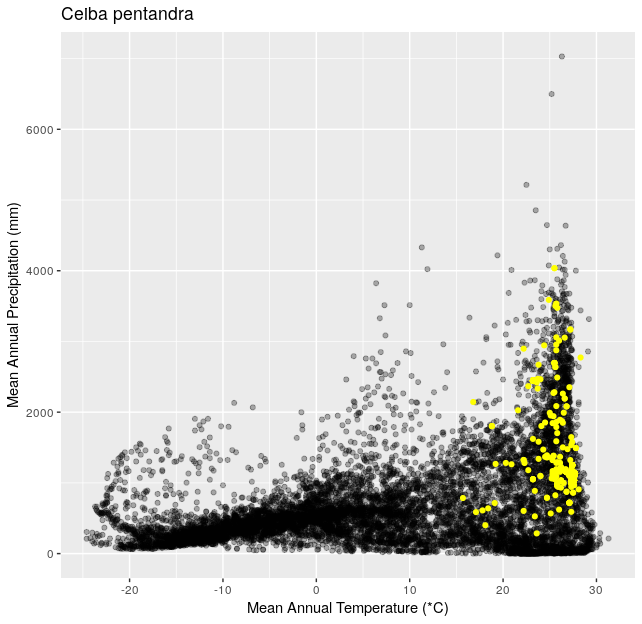
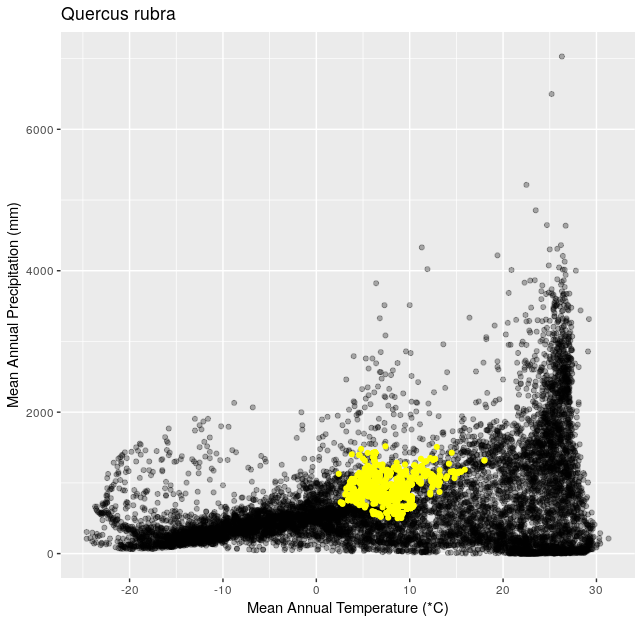
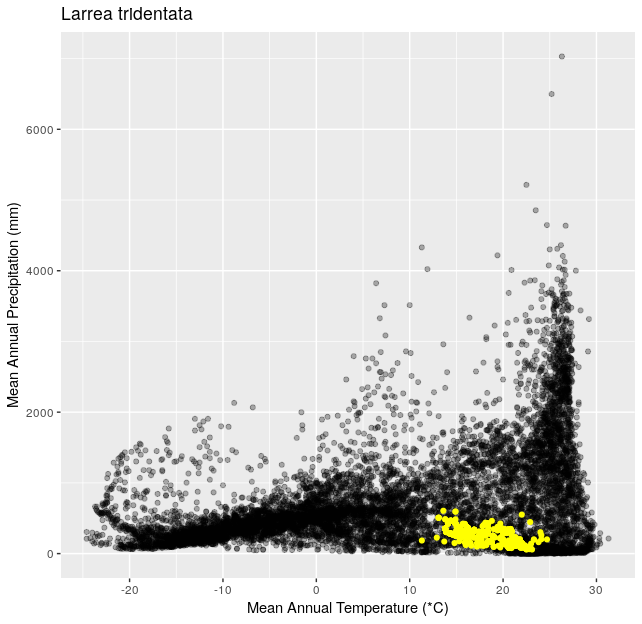
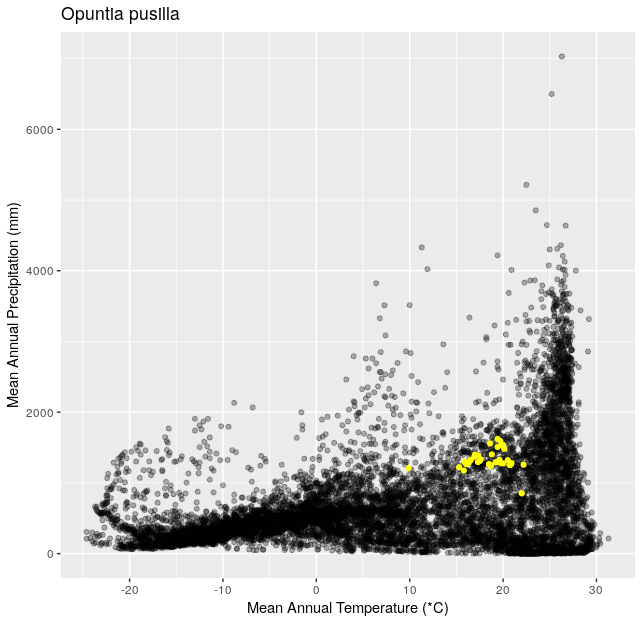
Climate Space Iteration (20 pts)
This is a follow up to Climate Space Rewrite.
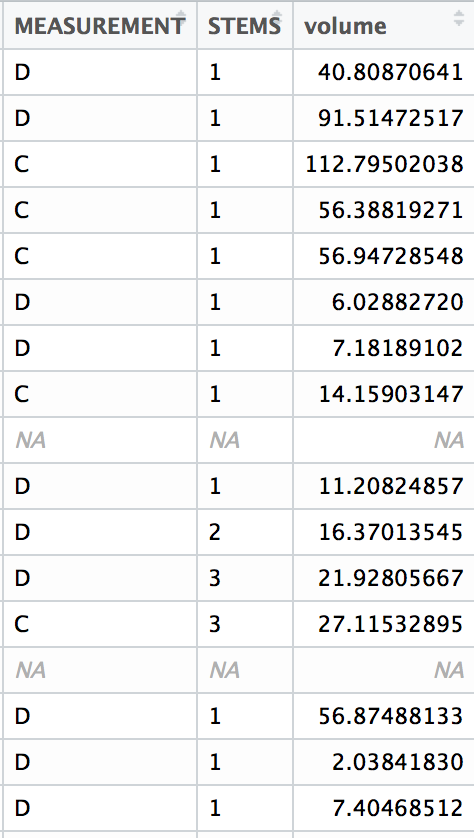
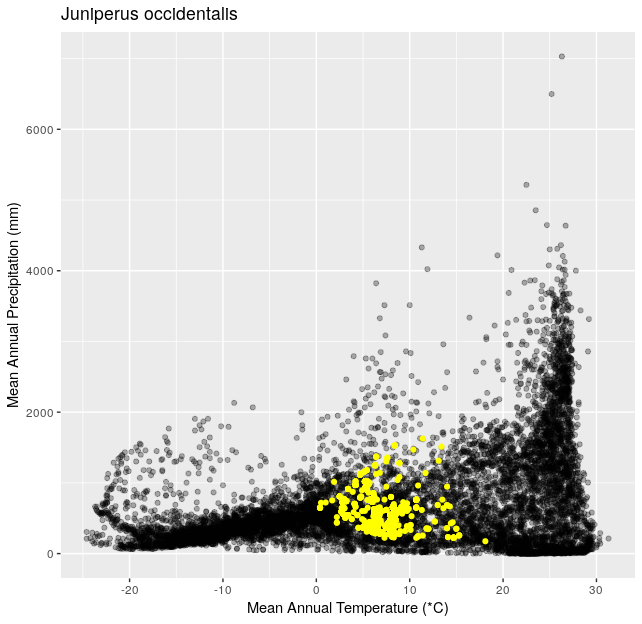
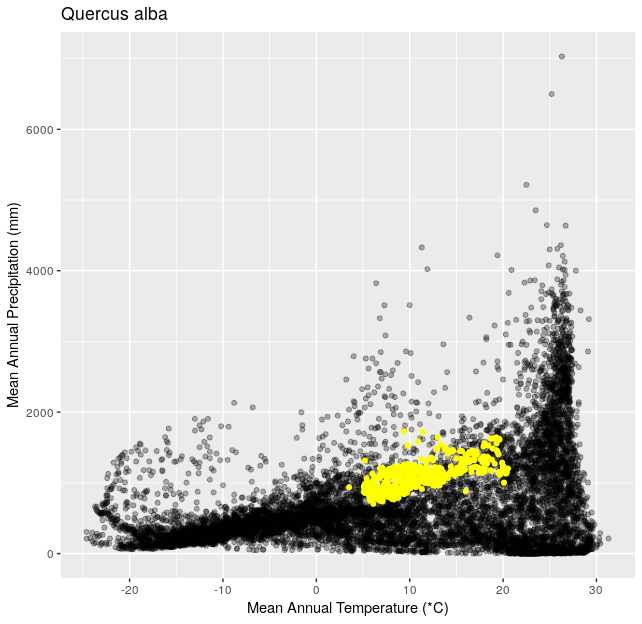
Using the functions you created in Climate Space Rewrite iterate over the following list of species to create one plot per species from the list. Include a title for each plot that is the species name using the
ggtitle()function. You can use any type of automated iteration that we’ve learned.[click here for output] [click here for output] [click here for output] [click here for output] [click here for output] [click here for output] [click here for output]species <- c("Juniperus occidentalis", "Quercus alba", "Picea glauca", "Ceiba pentandra", "Quercus rubra", "Larrea tridentata", "Opuntia pusilla")
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}